Dortmund, 4. August 2022
Kleinkinder, die kaum sitzen, geschweige denn krabbeln oder laufen können: Obwohl sie mit Spinaler Muskelatrophie (SMA) an einer seltenen Erkrankung leiden, häuften sich die Patient:innenfälle in der medialen Berichterstattung in den vergangenen Jahren. Der Grund für das Medieninteresse lag und liegt in den hohen Kosten einer 2020 in Europa zugelassenen Gentherapie für SMA: Eine Dosis Zolgensma® (Onasemnogen-Abeparvovec) kostet ca. zwei Millionen Euro und ist damit wesentlich teurer als das Alternativmedikament Spinraza® (Nusinersen). Zolgensma® soll bei Patient:innen unter zwei Jahren die fortschreitende Muskelschwäche nach einmaliger Gabe aufhalten. Um zu beurteilen, ob die SMA-Therapien anschlagen und wie sie verlaufen, kommen in der Praxis Biomarker als sogenannte Progressionsmarker ins Spiel. Am ISAS haben Wissenschaftler:innen beim Projekt »Gen- und Protein-Signaturen als GPS für Patienten mit neuromuskulären Erkrankungen« (koordiniert vom Universitätsklinikum Essen) ein Protein identifiziert, mit dessen Hilfe sich die SMA-Therapie optimieren lassen könnte.
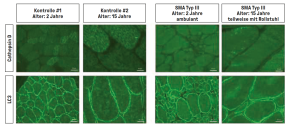
Die Mikroskop-Aufnahmen zeigen gefärbte Muskelbiopsien von sieben SMA-Patient:innen im Alter von 2 und 15 Jahren, die keine medikamentöse Therapie erhalten hatten. Ergebnis: Im Vergleich zu den Kontroll-Proben von gesunden Personen war das Protein Cathepsin D als einheitliches Muster in allen Proben herabreguliert. Als Kontrolle führten die Wissenschaftler:innen eine weitere Immunfärbung eines anderen Proteins, LC3, durch. Hierbei zeigten sich keine Veränderungen in den Muskelzellen von SMA-Patient:innen.
© Neuromuskuläes Labor der Klinik für Kinderheilkunde 1 am Universitätsklinikum Essen
Bei der Erbkrankheit SMA handelt es sich um eine Erkrankung der Nerven des Rückenmarks, der Motoneuronen. Ausgelöst wird SMA durch eine Mutation im Gen für das überlebende Motoneuron 1 (SMN1). Dieses Gen ist für die Produktion des SMN-Proteins »Survival Motor Neuron« verantwortlich, das die Gesundheit und normale Funktion der Motoneuronen aufrechterhält. Diese regulieren die Muskelaktivität, indem sie Signale aus dem zentralen Nervensystem senden – dem Teil des Nervensystems, das Gehirn und Rückenmark umfasst. Eine Degeneration der Motoneuronen führt zu einer allmählichen Abnahme der Muskelmasse und -stärke. Wenn es zu Schäden an den Neuronen kommt oder sie wie bei SMA absterben, setzt der Körper Strukturproteine, die Neurofilamente, im Blutkreislauf oder in der sogenannten Zerebospinalflüssigkeit (Gehirn und Rückenmark) frei. „Bei jungen SMA-Patient:innen ist die Anzahl der Neurofilamente erhöht. Während der Behandlung mit Spinraza® nehmen sie schneller ab als bei unbehandelten Patient:innen. Signifikante Veränderungen sind jedoch meist bei Kindern unter einem Jahr zu beobachten“, sagt Dr. Andreas Hentschel, Mitarbeiter der Arbeitsgruppe Translationale Analytik.
Cathepsin D als Progressionsmarker
Wie könnte ein Protein dabei helfen, die Therapie bei SMA zu optimieren? „Wir haben festgestellt, dass die Cathepsin-D-Werte in Liquor-Proben in unserer Kohorte von SMA-Patient:innen unter der Therapie mit Spinraza® abnehmen“, erläutert Hentschel.

Dr. Andreas Hentschel forscht unter anderem zu Biomarkern zur Kontrolle der Therapie und des Erkrankungsverlaufs bei SMA-Patient:innen.
© ISAS
Dieser Rückgang erschien laut dem Wissenschaftler bei allen SMA-Subtypen und Alterskategorien von zwei Monaten oder darüber. Zudem war die Abnahme bei Patient:innen, die eine positive motorische Reaktion auf die medikamentöse Behandlung zeigten, stärker ausgeprägt.
„Obwohl unsere Kohorte zu klein war, um Altersgruppen und SMA-Subtypen innerhalb der Gruppe der Therapie-Responder zu vergleichen, halten wir diese Ergebnisse für sehr vielversprechend. Sie deuten darauf hin, dass der Cathepsin-D-Spiegel als Biomarker auch bei älteren SMA-Patient:innen geeignet ist – in Kombination mit der Analyse des Proteins neurofilament light chain bei Jugendlichen oder als alleiniger Marker bei Erwachsenen“, resümiert Hentschel. Es seien weitere Validierungsstudien in größeren Kohorten und mit Serumproben erforderlich, um die Anwendbarkeit von Cathepsin D als Progressionsmarker für die verschiedenen SMA-Behandlungen zu überprüfen.
(Sara Rebein)
Das Projekt »Gen- und Protein-Signaturen als GPS für Patienten mit neuromuskulären Erkrankungen« wurde vom Land Nordrhein-Westfalen unter Einsatz von Mitteln aus dem Europäischen Fonds für regionale Entwicklung (EFrE) 2014-2020 »Investitionen in Wachstum und Beschäftigung« gefördert (Förderkennzeichen EFrE-0801301).